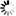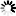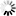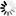
Sickle Cell Disease in Children
What is sickle cell disease?
Sickle cell disease is an inherited blood disorder characterized by defective hemoglobin (a protein in red blood cells that carries oxygen to the tissues of the body).
Sickle cell disease involves the red blood cells, or hemoglobin, and their ability to carry oxygen. Normal hemoglobin cells are smooth, round, and flexible, like the letter "O," so they can move through the vessels in our bodies easily. Sickle cell hemoglobin cells are stiff and sticky, and form into the shape of a sickle, or the letter "C," when they lose their oxygen. These sickle cells tend to cluster together, and cannot easily move through the blood vessels. The cluster causes a blockage in small arteries or capillaries and stops the movement of healthy, normal oxygen-carrying blood. This blockage is what causes the painful and damaging complications of sickle cell disease.
Sickle cells only live for about 10 to 20 days, while normal hemoglobin can live up to 120 days. Also, sickle cells risk being destroyed by the spleen because of their shape and stiffness. The spleen is an organ that helps filter the blood of infections and sickled cells get stuck in this filter and die. Due to the decreased number of hemoglobin cells circulating in the body, a person with sickle cell disease is chronically anemic. The spleen also suffers damage from the sickled cells blocking healthy oxygen carrying cells and typically infants in the first few years of life. Without a normal functioning spleen, these individuals are more at risk for infections. Infants and young children are at risk for life-threatening infections.
The most common variations of the sickle cell gene are:
-
Sickle cell trait. The child is carrying a single defective gene. Some of their hemoglobin is the destructive HbS, but they also have some normal hemoglobin, HbA. This is referred to as HbAS. Children with sickle cell trait are usually without symptoms of the disease. Mild anemia may occur and red cells tend to be small. Under intense stressful conditions, exhaustion, hypoxia (low oxygen), and/or severe infection, the sickling of the defective hemoglobin may occur and result in some complications associated with the sickle cell disease. Most children with the sickle cell trait lead normal lives.
-
Sickle cell anemia. The child has most or all of the normal hemoglobin (HbA) replaced with the sickle hemoglobin (HbS). This is referred to as HbSS. It is the most common and most severe form of the sickle cell variations. These children suffer from a variety of complications due to the shape and thickness of the sickled cells. Severe and chronic anemia is also a common characteristic for children with HbSS.
-
Sickle cell - hemoglobin SC disease. The child has one copy of both HbS and HbC. This is often referred to as HbSC. Hemoglobin C causes red blood cells, called target cells, to develop. Having just some hemoglobin C and normal hemoglobin, a person will not have any symptoms of anemia, but can develop it and eye and hip complications later in life. However, if the sickle hemoglobin S is combined with the target cell, some mild to moderate anemia occurs. These children often suffer some of the complications associated with HbSS, sickle cell disease, but to a milder degree. Vaso-occlusive crises (the flow of blood is blocked because the sickled cells have become stuck in the blood vessels), organ damage from repeated sickling and anemia, and high risk for infection are all similar traits for HbSS and HbSC.
-
Sickle cell - hemoglobin E disease. This variation is similar to sickle cell-C disease except that an element has been replaced in the hemoglobin molecule. This variation is most often also seen in Southeast Asia populations. Some children with hemoglobin E disease are without symptoms. However, under certain conditions, such as exhaustion, hypoxia, severe infection, and/or iron deficiency, some mild to moderate anemia may occur.
-
Hemoglobin S-beta-thalassemia. This involves an inheritance of both the thalassemia and sickle cell genes. The disorder produces symptoms of moderate anemia and many of the same conditions associated with sickle cell disease. While this disorder more often has milder symptoms than sickle cell disease, it may also produce exacerbations as severe as those of sickle cell disease.
All forms of sickle cell disease can exhibit the complications associated with the disease.
Who is affected by sickle cell disease?
Sickle cell disease primarily affects those of African descent and Hispanics of Caribbean ancestry, but the trait has also been found in those with Middle Eastern, Indian, Latin American, American Indian, and Mediterranean heritage.
It has been estimated that over 70,000 people in the U.S. are affected by sickle cell anemia and that 2 million people, and 10 percent of African-Americans have sickle cell trait. Millions worldwide suffer complications from sickle cell disease.
What causes sickle cell disease?
Sickle cell is an inherited disease caused by a genetic mutation. Genes are found on structures in the cells of our body called "chromosomes." There are normally 46 total, or 23 pairs, of chromosomes in each cell of our body. The 11th pair of chromosomes contains a gene responsible for normal hemoglobin production.
A mutation or error in this gene is what causes sickle cell disease. This mutation is thought to have originated in areas of the world where malaria was common, since people with sickle trait tolerate malaria better and have a reproductive advantage in areas with malaria. The sickle trait actually protects them from the parasite that causes malaria, which is carried by mosquitoes. Malaria is most often seen in Africa and in the Mediterranean area of Europe.
Sickle cell (HbSS) is a genetic disease. A person will be born with sickle cell disease only if two HbS genes are inherited—one from the mother and one from the father. A person who has only one HbS gene is healthy and said to be a "carrier" of the disease. They may also be described as having "sickle cell trait." A carrier has an increased chance to have a child with sickle cell disease. This type of inheritance is called autosomal recessive.
Autosomal means that the gene is on one of the first 22 pairs of chromosomes that do not determine gender, so that males and females are equally affected by the disease.
Recessive means that two copies of the gene, one inherited from each parent, are necessary to have the condition.
| |
| Click Image to Enlarge |
Children with sickle cell disease = S S (one in four, or 25 percent)
Children who are carriers of the gene like their parents = A S S A (two in four, or 50 percent have sickle cell trait)
Children who do not get the gene from either parent: A A (one in four, or 25 percent)
Once parents have had a child with sickle cell disease, there is a one in four, or 25 percent chance with each subsequent pregnancy, for another child to be born with sickle cell disease. This means that there is a three out of four, or 75 percent chance, that another child will not have sickle cell disease. There is a 50 percent chance that a child will be born with sickle cell trait, like the parents.
What are the symptoms of sickle cell disease?
The following is a list of symptoms and complications associated with sickle cell disease. Each child may experience symptoms differently. Symptoms and complications may include, but are not limited to, the following:
-
Anemia. This is the most common symptom of all the sickle cell diseases. In sickle cell disease, red blood cells are produced but then become deformed into the sickle shape, which causes red blood cells to lose their oxygen carrying capacity. The body subsequently becomes dehydrated, or with a fever. This sickle shape makes the cells stiff and sticky causing them to become stuck in the vessels, destroyed by the spleen, or simply die because of their abnormal function. The decrease in red blood cells causes anemia. Severe anemia can make a child pale and tired, and makes the child's ability to carry oxygen to the tissues more difficult. Healing and normal growth and development may be delayed because of chronic anemia.
-
Pain crisis, or sickle crisis. This occurs when the flow of blood is blocked to an area because the sickled cells have become stuck in the blood vessel. These are also called vaso-occlusive crises. The pain can occur anywhere, but most often occurs in the chest, arms, and legs. Painful swelling of the fingers and toes, called dactylitis, can occur in infants and children younger than age 3. Priapism is a painful sickling that occurs in the penis. Any interruption in blood flow to the body can result in pain, swelling, and possible death of the surrounding tissue not receiving adequate blood and oxygen.
-
Acute chest syndrome. This occurs when sickling is in the chest. This can be a life-threatening complication of sickle cell disease. It often occurs suddenly, when the body is under stress from infection, fever, or dehydration. The sickled cells stick together and block the flow of oxygen in the tiny vessels in the lungs. It resembles pneumonia and can include fever, pain, and a violent cough. Multiple episodes of acute chest syndrome can cause permanent lung damage.
-
Splenic sequestration (pooling). Crises are a result of sickle cells pooling in the spleen. This can cause a sudden drop in hemoglobin and can be life threatening if not treated promptly. The spleen can also become enlarged and painful from the increase in blood volume. After repeated episodes of splenic sequestration, the spleen becomes scarred, and permanently damaged. Most children, by age 8, do not have a functioning spleen either from surgical removal, or from repeated episodes of splenic sequestration. The risk of infection is a major concern of children without a functioning spleen. Infection is the major cause of death in children younger than age 5 in this population.
-
Stroke. This is another sudden and severe complication of children with sickle cell disease. The misshapen cells can block the major blood vessels that supply the brain with oxygen. Any interruption in the flow of blood and oxygen to the brain can result in devastating neurological impairment. Having had one stroke from sickle cell anemia, a child is 60 percent more likely to have a second and third stroke.
-
Jaundice, or yellowing of the skin, eyes, and oral mucosa. Jaundice is a common sign and symptom of sickle disease. Sickle cells do not live as long as normal red blood cells and, therefore, they are dying more rapidly than the liver can filter them out. Bilirubin (which causes the yellow color) from these broken down cells builds up in the system causing jaundice.
-
Priapism. An obstruction of the penis by sickle cells. If not promptly treated, it can result in impotence. Any and all major organs are affected by sickle cell disease. The liver, heart, kidneys, gallbladder, eyes, bones, and joints can suffer damage from the abnormal function of the sickle cells and their inability to flow through the small blood vessels correctly. Problems may include the following:
-
Increased infections
-
Leg ulcers
-
Bone damage
-
Early gallstones
-
Kidney damage and loss of body water in the urine
-
Eye damage
-
The symptoms of sickle cell disease may resemble other blood disorders or medical problems. Always consult your child's physician for a diagnosis.
How is sickle cell disease diagnosed?
In addition to a complete medical history and physical examination, diagnostic procedures for sickle cell may include blood tests, a complete family history, and results of newborn screening.
Many states now perform hemoglobinopathy testing (testing babies for abnormalities of hemoglobin) as part of the newborn screening blood tests that are routinely done. "State newborn screening" refers to a test done on every baby born in every state of the country within the first few days of life, to detect serious, life-threatening diseases. State laws require that babies be tested between 2 and 7 days of age, even if the baby seems healthy and has no symptoms of health problems.
Early diagnosis is essential in providing proper preventive treatment for some of the devastating complications of the disease.
A hemoglobin electrophoresis is a blood test that can determine if the child is a carrier of a specific sickle cell trait, or has any of the diseases associated with the sickle cell gene.
Treatment for sickle cell disease
Specific treatment for sickle cell disease and its complications will be determined by your child's physician based on:
-
Your child's age, overall health, and medical history
-
Extent of the disease
-
Your child's tolerance for specific medications, procedures, or therapies
-
Expectations for the course of the disease
-
Your opinion or preference
Early diagnosis and prevention of complications is critical in sickle cell disease treatment.
Treatment options may include, but are not limited to:
-
Pain medications (for sickle cell crises)
-
Drinking plenty of water daily (8 to 10 glasses) or receiving fluid intravenously (to prevent and treat pain crises
-
Blood transfusions (for anemia, and to prevent stroke; transfusions are also used to dilute the HbS with normal hemoglobin to treat chronic pain, acute chest syndrome, splenic sequestration, and other emergencies)
-
Penicillin (to prevent infections)
-
Folic acid (to help prevent severe anemia)
-
Hydroxyurea (A medication recently found to help reduce the frequency of pain crises and acute chest syndrome; it may also help decrease the need for frequent blood transfusions. The long-term effects of the medication are unknown.)
-
Bone marrow transplant (Transplants have been effective in curing some children with sickle cell disease; the decision to undergo this procedure is based on the severity of the disease and the availability of a suitable bone marrow donor. These decisions need to be discussed with your child's physician and are only done at centers that specialize in stem cell transplantation.)
Long-term outlook for a child with sickle cell disease
Several factors predict the long-term survival of a child with this disease including the following:
-
Type of disorder (whether a child has HbSS, HbSC, or other hemoglobin disorder)
-
Severity of the disease
-
Frequency of complications
-
Compliance with preventive regimens
The life expectancy has increased over the past 30 years and many individuals suffering with sickle cell disease can now live into their mid-40s and beyond. Advances in preventive care and new medications have reduced the life-threatening complications of sickle cell. However, it is still a severe, chronic, and sometimes fatal disease.
Related Questions
Related Articles









Health Calculators

-
Calculate your daily calorie intake depending on your weight loss goal.

-
It help you figure out whether you are overweight, underweight or...

-
This calculator gives the amount of calories your body would burn If...

-
It will display the number of calories you'll approximately burn...
Disclaimer - The content on this site is for informational purposes only. Always seek the advice of a qualified physician.
Copyright © 2025 sehat.com






